IRCCS Ospedale San Raffaele
Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS)
Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS)
Aperto 24 ore
0226431
Chiama oraL’eccellenza medico-scientifica dell’Ospedale San Raffaele nasce dalla triplice identità dell’istituto, un luogo dove ricerca, clinica e formazione universitaria interagiscono quotidianamente.
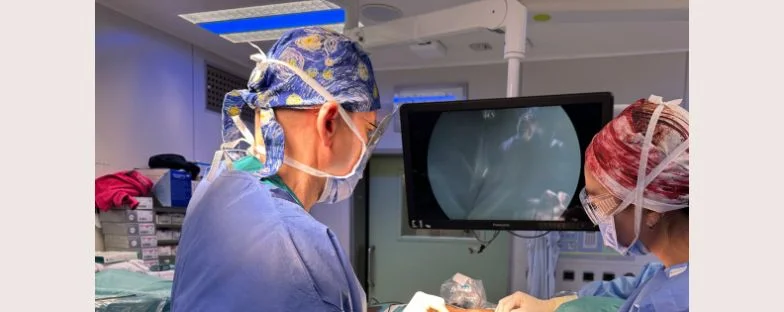
Dai robot chirurgici agli strumenti di imaging e radioterapia di precisione: all’Ospedale San Raffaele tecnologie all’avanguardia permettono diagnosi sempre più precoci e cure più personalizzate.
All’Ospedale San Raffaele le malattie vengono affrontate a 360°, grazie alla presenza di diversi specialisti che uniscono le loro competenze al servizio del paziente.
Si avvisa la gentile utenza che il nuovo orario della Farmacia e ritiro farmaci, a partire dal 1 luglio 2025, sarà dalle ore 8.30 alle 16.00 dal lunedì al venerdì. Il Medico prescrittore ospedaliero, in accordo con la Farmacia interna, è disponibile nel supportarvi nell’organizzazione del ritiro alternativo dei farmaci presso altre strutture pubbliche/ASST vicino al domicilio, continuando comunque a restare in cura presso l’Ospedale San Raffaele.
L’IRCCS Ospedale San Raffaele offre cure e assistenza in diverse specialità.








Trova lo specialista più indicato in area chirurgica, area medica e servizi clinici dell'Ospedale San Raffaele.


Screening, approfondimenti e attività per grandi e bambini: il 29 giugno dalle ore 10, gli specialisti dell'Ospedale San Raffaele ti aspettano alla Società Umanitaria di Milano per celebrare la salute e il benessere.

Grazie alla profonda integrazione tra ricerca di base, traslazionale e clinica, e alle numerose collaborazioni con i principali centri di ricerca internazionali, l’Ospedale San Raffaele è leader in molti campi di ricerca e offre la migliore assistenza ai suoi pazienti.



/resolutions/res-c160x160/Banner-genos_OSR_449x328-(2).jpg)





È dotato di un suo organico e si avvale della stretta cooperazione con tutte le specialità cliniche dell’ospedale.
Un punto prelievo è attivo dal lunedì al sabato dalle ore 7.00 alle 11.00.
Esegue circa 6.500.000 analisi di laboratorio all’anno con strumentazioni d’avanguardia.
Sono presenti 4 parcheggi all’interno e nelle aree limitrofe dell’ospedale. È presente anche una stazione di ricarica per auto elettriche.
È presente un servizio di portineria 24 ore su 24.
Per qualunque informazione gli utenti possono fare affidamento a un ufficio dedicato.
All’interno dell’ospedale è presente una palestra per attività di riabilitazione.
Su richiesta è possibile mettere a disposizione dei pazienti camere con televisione.
Il nostro istituto offre su richiesta assistenze dedicate in lingua.
Sono presenti diversi bar e punti ristoro all’interno del campus ospedaliero.
Sia degenti che visitatori possono utilizzare il servizio mensa.
All’interno del campus sono presenti diverse aree fornite di distributori automatici.
Sono presenti due Cappelle dove si svolgono quotidianamente le funzioni religiose
È prevista la presenza di un sacerdote e, per i fedeli di altre confessioni religiose, è possibile richiedere la presenza di un ministro.
È presente un servizio ristorante.
È possibile prenotare il servizio di parrucchiere o barbiere rivolgendosi al capo sala o al personale infermieristico.
Sono attivi due sportelli bancomat all’interno dell’ospedale.
La nostra struttura è dotata di una rete Wi-Fi usufruibile da utenti esterni e interni.
È presente un punto vendita di giornali, riviste e libri.
L’intero campus si trova all’interno di uno spazio verde e curato.
Un servizio navetta collega velocemente l’ospedale con la fermata M2 Cascina Gobba.
Sono molte le associazioni di volontariato attive all’interno del nostro ospedale.
È presente un piccolo ma fornito supermercato nella galleria dei negozi.
È presente una parafarmacia con articoli ortopedici.
Fiorista, tintoria, abbigliamento, gioielleria, giocattoli, Lindl, agenzia assicurativa.
È attiva una collaborazione con i cani della SICS.